

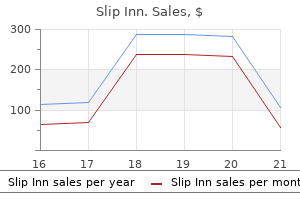
Slip Inn dosages: 1pack
Slip Inn packs: 10 caps, 20 caps, 30 caps, 60 caps, 90 caps, 120 caps, 180 caps

1pack slip inn buy
Hyperinsulinism in short-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency reveals the importance of beta-oxidation in insulin secretion herbs cooking purchase slip inn 1pack. Familial hyperinsulinemic hypoglycemia with a mutation in the gene encoding short-chain 3-hydroxyacyl-CoA dehydrogenase herbals vaginal dryness cheap slip inn 1pack. The cblD defect causes eith isolated or combined deficiency of methylcobalamin and adenosylcobalamin synthesis. Mechanism of hyperinsulinism in short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency includes activation of glutamate dehydrogenase. Of two affected siblings within the initial report of Gibson and colleagues in 2000 [1], the first manifested neurologic abnormalities following a probable ischemic/hypoxic event at three days of age. His sister, recognized prenatally, had been fully asymptomatic by the time of follow-up report [2], and numerous other patients have been asymptomatic, significantly these recognized by neonatal screening [2�5]. The key to the metabolic abnormality was the excretion of 2-methylbutyrylglycine within the urine and an elevated level of 2-methylbutyrylcarnitine in the blood. By 12 months of age, he was behind in visible, motor, and cognitive skills and carried a prognosis of athetoid cerebral palsy. In one other family [2, 3], the patient was a three-year-old product of a consanguineous mating who had hypotonia and impaired motor improvement. Among ethnic Hmongs, eight patients have been discovered on expanded new child screening to have 2-methylbutyrylglycinuria and a distinct mutation [4]. Except for mild muscular hypotonia observed at six months of age, all had been asymptomatic. Of two different consanguineous patients, one had attention-deficient disorder, and one convulsive disease and developmental delay. It appears that sufferers with this disorder could also be asymptomatic, however neurologic illness could additionally be a feature. Interelations of the S and R pathways of isoleucine catabolism the gene incorporates 11 exons [7]; its open studying body of 1. Mutational evaluation in the first affected person revealed a C778T substitution in the coding area which led to the substitution of a phenylalanine for leucine at amino acid 22 [1]. Mutational evaluation revealed homozygosity for a G1228A transition within the second affected person and his mom [3]. Mutational evaluation within the Hmong patients yielded a homozygous A1165G mutation, which additionally led to skipping of exon 10. Incubation of to 2 13C-isoleucine with L-carnitine in intact cultured fibroblasts led to accumulation of isotope in C5-acylcarnitine. In the second affected person [3], the exercise of 2-methylbutyrylCoA dehydrogenase in fibroblasts was 10 % of management. Defective activity was additionally demonstrated by expressing the abnormal gene merchandise in E. Many of those reported have been found through applications of expanded newborn screening. Elevated C5 acylcarnitine may be documented by analysis of the plasma, as properly as of dried blood on filter paper. Chiral willpower of 2-methylbutyric acid indicated that 40�46 p.c was in the type of the R isomer in patients and in controls. Keto-enol tautomeric racemization following or enamine tautomerization throughout, transamination is the source of alloisoleucine (Chapter 19). Ethylhydracrylic acid excretion in increased amount may also be noticed in ketosis [12], in 3-oxothiolase deficiency [13], in 2-methyl-3-hydroxybutyrylCoA dehydrogenase deficiency [14], in propionic acidemia [15], and methylmalonic acidemia, all defects in steps of the S pathway. It may also be present in ethylmalonic encephalopathy, hydroxyisobutyric aciduria, and in Barth syndrome. Prenatal prognosis was completed [1] by analysis of 2-methylbutyrylglycine in amniotic fluid (0. A protein intake of 1�4 g/kg together with carnitine of 71 mg/kg led to a traditional excretion of 2-methylbutyrylglycine [1]. Isolated 2-methylbutyrylglycinuria caused by short/branched-chain acyl-CoA dehydrogenase deficiency: identification of a brand new enzyme defect, resolution of its molecular basis, and evidence for distinct acyl-CoA dehydrogenases in isoleucine and valine metabolism. Prospective diagnosis of 2-methylbutyryl-CoA dehydrogenase deficiency in the Hmong population by new child screening using tandem mass spectrometry. Stable isotope dilution evaluation of n-hexanoylglycine, 3-phenylpropionylglycine and suberylglycine in human urine using chemical ionization gas chromatography/mass spectrometry selected ion monitoring. Evaluation of urinary acylglycines by electrospray tandem mass spectrometry in mitochondrial energy metabolism defects and organic acidurias. Hydroxycarboxylic and oxocarboxylic acids in urine: products from branched-chain amino acid degradation and from ketogenesis. Propionyl-CoA carboxylase deficiency with overflow of metabolites of isoleucine catabolism in any respect ranges. Patients with this type of the dysfunction have overwhelming illness within the neonatal period that has been uniformly fatal. The name was employed to distinguish the illness from the glutaric aciduria as a end result of faulty exercise of glutaryl-CoA dehydrogenase (Chapter 9) that had been reported one year earlier by Goodman and colleagues [2]. Organic acid evaluation revealed the buildup of all kinds of natural acids, including lactic, isovaleric, and ethylmalonic acids, as well as glutaric acid. There is generalized defect in the activity of a minimum of 9 acyl CoA dehydrogenases [3]. The fundamental molecular defect is within the mitochondrial transport of electrons from the acylCoAs to ubiquinone (CoQ10) of the primary electron transport chain [5�7]. There seems to be a much larger incidence in the Turkish inhabitants, > 1:20,000 [33]. The first affected person was described as having a "very unpleasant sweaty-feet odor" [1]. This is the consequence of an excess of a selection of short-chain, volatile natural acids. A number of these patients have been described as pale [1, 34, 35] and one had macrocytic anemia and a hemoglobin concentration of 9. Many have had convulsions according to the diploma of depression of the blood glucose. They embody a high forehead, depressed nasal bridge, and a short anteverted nose. Muscular defects of the abdominal wall have occurred, as properly as genital defects, similar to hypospadias and chordee. He had a excessive brow, depressed nasal bridge, brief nostril with anteverted nares, a long philtrum and micrognathia. He had a low, incompletely rotated, low-s ears with a lowered anthelix, semilunar folds beneath the eyes and three umbilical vessels. One of our patients additionally had an interventricular septal defect and three umbilical vessels.
Syndromes
- Itchiness
- Not being able to control behavior
- Temporary paralysis of the bowel (paralytic ileus)
- Peas and beans (legumes), seeds
- Breathing difficulty (from breathing in chemical)
- Drooling, which may often begin before teething starts
- After the area is dry, a surgical cut is made. The skin is pulled back until the skull is seen.
- Absent or decreased tears
- Live in climates with little exposure to sunlight
Slip inn 1pack effective
Akt activation inhibits p53 proapoptotic signaling via the phosphorylation and stabilization of Mdm2 herbs mopar 1pack slip inn discount overnight delivery, an E3 ubiquitin ligase that rupam herbals slip inn 1pack order without a prescription, when activated, targets p53 for degradation. FimA is a pilus protein that in the cytosol of infected cells antagonizes cytochrome c launch and subsequent caspase-3 activation (96). This prevents hexokinase dissociation from the mitochondrial membrane, an event that triggers Bax translocation to the outer mitochondrial membrane, the formation of Bax/Bak mitochondrial outer membrane pores, loss of membrane integrity, and release of cytochrome c. Since the infectious dose of Shigella could be as low as 10 to one hundred bacteria, safeguarding its replicative niche is of particular importance early during the an infection course of in order to enhance the bacterial load. Shigella evades host cell dying by obstructing the activation of pro-death pathways and by activating pro-survival pathways. The inhibition of epithelial cell demise is spectacular, as Shigella can replicate to excessive numbers (>100 bacteria) with out lysing the epithelial cell, which is in stark distinction to the fast cell demise induced in macrophages. OspC3 binds to the p19 subunit of caspase-4 and thereby inhibits the assembly of the active caspase-4 tetramer, consisting of two p19 and two p10 subunits. Shigella thereby makes use of the activation of pro-survival pathways to counteract necrotic and apoptotic cell death. In addition to obstructing host cell death, Shigella also preserves its intracellular niche inside the epithelial layer by selling the adhesion of infected cells to the basement membrane and stopping cell division. To forestall division, and the accompanying discount of adhesion to neighboring cells and the lamina propria, the Shigella effector IpaB can mediate cell cycle arrest by interfering with the binding of the anaphase-promoting complex inhibitor Mad2L2 to Cdh1 (103), which is part of the anaphase-promoting complicated and is involved in preventing mitosis by suppressing mitotic cyclins needed to enter mitosis. Lack of binding of Mad2L2 to Cdh1 retains Cdh1 constitutively active and prevents cells from undergoing mitosis. Through these a number of mechanisms, Shigella safeguards its replicative niche and ensures adequate time to replicate extensively within the host cell and to spread to neighboring cells. Transcriptional profiling and world proteomic evaluation of Shigella throughout cytosolic growth have given a good overview of the metabolic adjustments that accompany Shigella adaptation to the cytosolic replicative area of interest. In contrast to its extracellular development, Shigella adapts to the low-oxygen setting of the epithelial cytosol by downregulating enzymes concerned in oxidative respiration and increasing expression of enzymes concerned in glycolysis and mixed-acid fermentation (105). Shigella additionally increases fructose and mannose importers in addition to the dipeptide transporter DppA to scavenge vitamins from the host. Due to the inherent toxicity of iron, the host limits intracellular iron concentrations and sequesters free iron by expressing iron-binding proteins. Iron is an important micronutrient for Shigella growth, and Shigella can capture intracellular iron through a number of routes, including the expression of siderophores and uptake of heme or ferric and ferrous iron by way of devoted iron transport methods, which Shigella upregulates in response to the low iron accessibility in the cytosolic milieu (106). Analysis of auxotrophic mutants of Shigella additionally indicates that Shigella has entry to diverse amino acids in adequate quantities, whereas availability of host fatty acids, purine nucleosides, and some amino acids, similar to asparagine and proline, is limiting. While Shigella obtains a considerable quantity of biomass by way of uptake of nutrients, metabolite mass spectrometry utilizing 13C glucose isotype tracking revealed that Shigella derives its vitality virtually solely from pyruvate during cytosolic growth in HeLa cells (107). Pyruvate is a very abundant metabolite in HeLa cells, as these most cancers cells mostly rely on glycolytic pathways rather than the oxidative tricarboxylic acid cycle for vitality production. Shigella metabolizes pyruvate into acetate using the low-yield however high-speed acetate 30 pathway. Rather, Shigella efficiently makes use of the waste product of the host power production to power its replication. Shigella infection of epithelial cells does, nonetheless, activate metabolic stress responses, as metabolic labeling of de novo protein synthesis shows a world shutdown of host protein synthesis, significantly early within the first two hours during an infection (108). To assist the metabolic needs of its rapid replication, Shigella might profit from the amino acid assets made available via the reduction in general host translation and stress-induced stimulation of amino acid metabolism. Both pathways use the activation of presynthesized transcriptional activators, permitting a speedy transcriptional response to dangerous stimuli. This contains targeted inhibition of factors concerned in proinflammatory pathways, interference in transcriptional activation of proinflammatory cytokines, inhibition of the endomembrane 2. OspF has a phosphothreonine lyase activity, a uncommon enzymatic exercise that irreversibly dephosphorylates threonine by way of beta elimination (125). Notably, phosphoproteomics showed that OspF affects the phosphorylation standing of a quantity of hundred host proteins, highlighting the extensive impact a single effector can have, immediately and indirectly, on host signaling networks (128). Vesicular trafficking, required for cytokine secretion, can be subverted by Shigella with the injection of the effectors IpaJ and VirA, and likely IcsB, as IcsB targets and modifies quite a few host proteins involved in vesicular 32 trafficking (76). The activity of IpaJ, and to a lesser degree VirA, thereby leads to a putting fragmentation of the Golgi membrane. Connexin hemichannels are gated pores fashioned on the plasma membrane by way of the meeting of six connexins into a hoop structure. Connexin channels enable the flow of ions and signaling molecules both to the extracellular milieu in the case of hemichannels or between neighboring cells via the stacking of hemichannels at sites of cell-cell contact. The multiple mechanisms utilized by Shigella and the various host pathways focused throughout an infection of epithelial cells to restrict inflammation clearly show the essential function innate immune evasion performs in selling infection. The primary portal of entry for Shigella throughout its crossing of the epithelial barrier is the specialised lymphoid follicle-associated epithelium, bringing Shigella into early contact with both innate and bought immune cells. At the identical time, Shigella can migrate to the draining mesenteric lymph nodes using the host lymphatic system. Shigellosis is characterized by massive T and B cell demise in rectal biopsy samples and by poor long-lived B cell immunity, requiring a number of infections for the host to turn out to be extra proof against an infection. Shigella is therefore effective in inhibiting the technology of long-lived immunity and sure does this via several mechanisms. In in vivo fashions, Shigella has been demonstrated to invade each T and B cells, while in vitro, Shigella is capable of invading B cells and activated, but not unactivated, T cells (44, one hundred thirty five, 136). Mechanistic insights were unraveled in vitro, where Shigella inhibited T cell motility in path of a chemokine attractant in an IpgD-dependent manner (135). Conversely, Shigella interactions with B cells result in each necrotic and apoptotic cell demise relying on the interaction sample (44). While invasion of B cells leads to necrotic cell demise, noninfected B cells primarily bear apoptosis. Induction of B cell dying in both invaded and noninvaded cells antagonizes antibody-mediated immune responses and likely contributes to the poor priming of B cell immunity to Shigella an infection. However, the context of timing and cell specificity are additionally necessary issues. Thus, Shigella benefits from irritation early throughout an infection and actively promotes the proinflammatory pyroptosis of macrophages whereas using a multitude of effectors to damp the host inflammatory responses in epithelial cells. Having developed to utilize the host cell cytosol as its major replicative area of interest, Shigella targets quite a few host cell processes to guarantee its personal survival and the integrity of the infected cell. These host cell processes could also be focused by a single effector or by many, and an effector might have one or multiple mobile targets or an enzymatic exercise that can affect a variety of processes. In addition, whereas most investigations have centered on elucidating effector function in infected cells, latest advances in our understanding of interplay of Shigella with cells of the adaptive immune system have already highlighted the function that some Shigella effectors can play independently of cell invasion to subvert host cell perform. This new "kiss and run" model provides a further layer of complexity to the study of Shigella pathogenesis and, like many other areas of Shigella pathogenesis, has strongly benefited from the development of novel strategies and new strategies.
Slip inn 1pack discount online
Deep tendon reflexes are exaggerated herbs collinsville il 1pack slip inn generic with amex, and there could additionally be ankle clonus and constructive Babinski responses herbs lung cancer buy cheap slip inn 1pack on line. Neurologic deterioration is progressive and could also be rapid following intercurrent sickness and results in terminal coma and dying, typically within the first to fourth 12 months [5, 7, 9]. Hypotonia, head lag, and delayed growth have been noted as early as three to 4 months [5�7, 9]. She had epicanthal folds, upslanting palpebral fissures, an upturned nostril, and depressed nasal bridge. Patients also have episodic showers of petechiae, typically related to an infection. At two months, there was diminution of mature megakaryocytes regardless of an elevated variety of younger megakaryocytes. She presented at birth with severe thrombocytopenia unresponsive to cortisone or immunoglobulins [5]. From the second yr of life, the affected person needed an rising variety of transfusions of platelets and pink blood cells. There was no proof for an immunologic abnormality, nor were there abnormalities of bleeding, clotting, or platelets. A markedly elevated level of plasminogen activator inhibitor-1 has been encountered [9]. Terminal occasions in two patients appeared to be pulmonary edema and one had cerebral edema. Between attacks, the blood concentrations of lactate and pyruvate remained excessive, and metabolic acidosis is compensated. On electron microscopy, there were mildly increased numbers of pleomorphic subsarcolemmal mitochondria [9 and G. There was a relative sparing of neurons and pallor of the background parenchyma that was quite prominent within the substantia nigra. Note progressive atrophy and encephalomalacia of putamen, caudate, lentiform nuclei, and periventricular white matter with cystic adjustments. Taken collectively, the clinical course of ethylmalonic encephalopathy is characterized by medical heterogeneity; Pigeon and colleagues reported different clinical courses even in monochorionic twins [17]. Since the preliminary report, only about forty circumstances of ethylmalonic encephalopathy have been described worldwide, suggesting that this situation is an ultra-rare autosomal recessive disorder. Most sufferers with ethylmalonic encephalopathy have been, with a couple of exceptions, of Mediterranean [5, 6, eight, 14] or Arabic [7] descent. However, the actual incidence of this situation may have been significantly underestimated as a outcome of the biochemical phenotype may be incorrectly attributed to different metabolic disorders, particularly defects of the mitochondrial electron-transfer pathway. Some of these sufferers had been proven and more may have been ethylmalonic encephalopathy. Patients reported have been Yemeni, Italian, Egyptian, Native American, and Hispanic-American. The main metabolic abnormality is the excretion of ethylmalonic acid within the urine. In some patients, the excretion of methylsuccinic acid was also elevated, but in others it was not, and ranges had been as high as 266 mmol/mol creatinine (normal <12) in a single affected person. Adipic aciduria was not current, although in one patient a level as excessive as 334 mmol/mol creatinine was recorded. Tandem mass spectrometry of the blood and urine showed an elevation in C4 and C5 carnitine esters and excretion elevated after therapy with carnitine. Acylglycine excretions have been increased, including butyrylglycine and isobutyrylglycine [6, 13], in addition to 2-methylbutyrylglycine and isovalerylglycine [13]. There was marked endothelial proliferation of capillaries and an increase in the variety of capillaries (H&E, �500). In these studies, the oxidation of 14C-butyrate was regular, according to our findings on triglyceride loading. In our affected person, loading with methionine was followed by a rise in excretion of ethylmalonic acid of 1. A relationship between this syndrome and the metabolism of sulfur amino acids was instructed by Duran and colleagues [19] who found increased excretion of inorganic thiosulfite and an absence of detectable sulfite. They also reported two sulfur-containing acidic amino acids, S-sulfocysteine and S-sulfothiocysteine, each of which may be shaped nonenzymatically from thiosulfate and cysteine. The improve in excretion of ethylmalonic acid in our patient following methionine is according to these observations. Homoserine is converted to 2-oxobutyric acid which might be a source of ethylmalonic acid. Cysteine is converted to 2-mercaptopyruvic acid which is metabolized to pyruvic acid and thiosulfate and in the end sulfate [20]. Ethylmalonic acid could be a product of isoleucine metabolism through the R pathway after racemization of 2-oxo-3-methylvaleric acid, the precursor of alloisoleucine, from the S to the R type, which is then convertible to 2-methylbutyryl CoA, 2-ethyl-3-hydroxypropionyl CoA, and ethylmalonic semialdehyde after which to ethylmalonic acid. It has been proven that methylsuccinic acid is formed from ethylmalonic acid in bacteria [22]. The gene for this illness was discovered through homozygosity mapping to reside in chromosome 9q13 close to the midpoint of the lengthy arm [8]. A constitutive knockout mouse model was constructed, which demonstrated impaired progress, decreased motor activity, and early dying. Thiosulfate concentrations have been discovered to be several-fold greater than management, and sulfite was undetectable. Hydrogen sulfide is a volatile molecule which has important toxic results, but can also serve in some intracellular regulatory functions [24]. The main portion of bodily H2S arises from bacterial metabolism, but there can also be appreciable endogenous production. H2S is also produced in vascular endothelium via the actions of 3-mercaptopyruvate sulfurtransferase and cysteine aminotransferase [26], the place it could act as a clean muscle relaxant [27]. There have been deletions, together with heterozygous [31] and homozygous [32] deletions of exon 4. There have been at least 4 haplotypes in six unrelated cases which contain the mutation c. R163W), in addition to two different missense mutations at R163 [29], indicating a possible mutational hotspot. Treatment with riboflavin, carnitine, glycine, and vitamin E References 785 have been without evident effect. In our patient [9], a food plan restricted in methionine led to a decrease in excretion of ethylmalonic and methylsuccinic acids and normalization of concentrations of lactic acid and bicarbonate, but the disease was relentless, and she died at eleven months of age. Early trials of broad-spectrum enteral antibiotic (metronidazole at round 30 mg/kg per day) and an agent to increase glutathione (N-acetylcysteine at around 100 mg/kg per day) showed marked profit by means of the cutaneous manifestations and, in some cases, additionally in neurologic symptoms [31�33]. Progressive deadly pancytopenia, psychomotor retardation and muscle caritine deficiency in a child with ethylmalonic aciduria and ethylmalonic acidemia.
Slip inn 1pack buy visa
There may be patchy ossification of the frontal bones and small plaques of parietal or occipital bone herbals for blood pressure slip inn 1pack cheap without a prescription. Roentgenographic examination [3 herbs de provence buy discount slip inn 1pack line, four, 16] reveals generalized rarefaction of the skeleton. The roentgenographic abnormalities of this disease distinguish it from all different disorders of bone. These sufferers could additionally be stillborn or die inside hours of start of respiratory problem, or shortly later of pulmonary an infection. A reasonably extreme or infantile kind could present within the first months of life. There have been no survivals in patients with hypophosphatasia presenting with medical manifestations prior to the end of the primary six months. The cranial sutures are extensive, and a bulging anterior fontanel and outstanding scalp vein usually develop. In sufferers with hypophosphatasia first seen after the seventh month of life and throughout childhood, the illness could additionally be less extreme. Convulsions, brain harm, or even demise could additionally be problems in the absence of surgical decompression. These patients may have retarded progress and increased susceptibility to an infection. There could additionally be options suggestive of rickets with costochondral beading and widening of the ends of the bones, in addition to the bowing deformity but, on X-ray, the ends of the bones have a notched appearance very completely different from the cupping of rickets and extra like that of a metaphyseal dysplasia. The incidence of subperiosteal new bone formation additionally distinguishes this picture from that of rickets [23]. In fact, some have been observed in whom untimely lack of the anterior deciduous teeth was the only proof of disease [18, 19, 24]. These and different sufferers might have bone pains, and occasional fractures remain a half of life. Painful ft may point out recurrent and poorly therapeutic metatarsal stress fractures. Some patients have presented in maturity, however give a history of rickets in childhood [6]. Recurrent arthritis and widespread calcification of articular cartilages was described in a 51-year-old woman with hypophosphatasia [18, 34]. The term pseudohypophosphatasia [18, 26] refers to patients with otherwise typical hypophosphatasia in whom Treatment 799 the extent of alkaline phosphatase in the plasma is regular. The fact that sufferers with pseudohypophosphatasia and with hypophosphatasia could also be present in the identical kindred indicates that this represents the vagaries of enzyme degree in the serum somewhat than a distinct illness entity [28�32]. The main criterion for prognosis of hypophosphatasia is an absent or extraordinarily low exercise of alkaline phosphatase in serum [39]. Alkaline phosphatase exercise is normal in gut and placenta in these patients [40], indicating that the isozyme in these tissues is genetically different from that of plasma, bone, and different tissues. Alkaline phosphatase is believed to be concerned in the deposition of mineral crystals by the hydrolysis of pyrophosphate and other phosphate esters. This amino acid is a useful marker for genetic studies and the detection of heterozygosity. However, its presence in the urine in different metabolic ailments of bone [44] makes it much less helpful in analysis. Ranges in mol/g creatinine are: 83�222 in those lower than 15 years of age, 42�146 in 16 to 30 years; 38�155 in 31 to 41 years and 48�93 in older individuals [44]. The concentrations of inorganic pyrophosphate in plasma and urine have been reported [46] to be elevated. There are extensive, irregular zones of proliferative cartilage and an absence of calcification of the osteoid. Variability and errors in the assay of alkaline phosphatase are so widespread that excretion of phosphoethanolamine is probably a more dependable method for the research of households. Prenatal prognosis has been achieved by the ultrasonographic look of the fetal head at sixteen weeks [47]. Prenatal diagnosis can be accomplished by the measurement of alkaline phosphatase activity in cultured amniocytes [48]. Family studies point out clearly that the different medical phenotypes symbolize totally different illness entities. Certainly, the extreme neonatal illness and the delicate juvenile illness never occur collectively in a kindred [27]. A form of hypophosphatasia has been reported [49] in which transmission is autosomal dominant. Clinical options had been untimely loss of tooth, bowed legs, and a crushed silver appearance to the roentgenogram of the cranium. It is obvious that the classic delicate hypophosphatasia, including asymptomatic people who have phosphoethanolaminuria and some with odontohypophosphatasia have dominant expression of mutation on a single allele [7, 50�52]. E174K mutation is relatively frequent in Caucasians, occurring in 31 p.c of individuals with delicate hypophosphatasia [56]. The heterozygous state can usually be demonstrated by measuring the serum exercise of alkaline phosphatase [3]. However, it often is inconceivable to distinguish carriers from patients on the basis of activity of alkaline phosphatase. The traditional measures are employed for the management of fractures and skeletal deformities. Nonsteroidal anti-inflammatory agents could also be useful for bone pain and stress fractures. Bone marrow transplantation has been with out impact in extreme perinatal hypophosphatasia, but transplantation of allogeneic mesenchymal stem cells cultured underneath osteogenic circumstances and osteogenic constructs made by growing cells on porous hydroxyapatite ceramic yielded proof of improved mineral density of bone and de novo bone formation around the constructs [58]. Enzyme replacement therapy has been underneath scientific investigation in infants and kids. The human gene was bioengineered by extending the C terminus and addition of a deca-aspartate sequence to target mineralizing tissue. Clinical trials reported to date have proven improvement in roentgenographic look, motor perform, energy, and agility [60]. Different missense mutations on the tissue-nonspecific alkaline phosphatase gene locus in autosomal recessively inherited forms of gentle and extreme hypophosphatasia. Identification of novel missense mutations Phe310Leu and Gly439Arg) in a neonatal case of hypophosphatasia. Analysis of localization of mutated tissue-nonspecific alkaline phosphatase proteins related to neonatal hypophosphatasia utilizing green fluorescent protein chimeras. Glu274Lys/Gly309Arg mutation of the tissue-nonspecific alkaline phosphatase gene in neonatal hypophosphatasia related to convulsions. Rickets, deficiency of "alkaline" phosphatase exercise and untimely loss of teeth in childhood. Childhood hypophosphatasia and the premature loss of teeth: A medical and laboratory study of seven circumstances. Hypophosphatasia: Screening and family investigations in an endogamous Hungarian village. Hypophosphatasia related to calcium pyrophosphate dihydrate deposits in cartilage.
Order slip inn 1pack fast delivery
Natural historical past and clinical management of emphysema in patients with and without alpha-1-antitrypsin inhibitor deficiency wonder herbals order slip inn 1pack without prescription. Pathophysiology of the pulmonary circulation in emphysema related to alpha1-antitrypsin deficiency herbals for horses slip inn 1pack generic free shipping. Symptomatic pulmonary emphysema in childhood associated with hereditary alpha1-antitrypsin and elastase inhibitory deficiency. A comparison between clinical roentgenologic functional and morphologic criteria in chronic bronchitis emphysema bronchial asthma and bronchiectasis. Effect of cigarette smoking on the pulmonary operate of children and adolescents. Membranoproliferative glomerulonephritis in childhood cirrhosis related to alpha-1-antitrypsin deficiency. Association of extreme rheumatoid arthritis with heterozygosity for 1-antitrypsin deficiency. Liver disease in alpha-1-antitrypsin deficiency detected by screening of 200000 infants. Discovery of a ninth allele belonging to the system of inherited variants of serum 1�antitrypsin. Liver injury in a neonate with alpha-1-antitrypsin deficiency as a end result of phenotype PiZ Null (Z �). Assignment of the 1antitrypsin gene and a sequence-related gene to human chromosome 14 by molecular hybridization. Detection of point mutations in amplified single copy genes by biotin-labelled oligonucleotides: Diagnosis of variants of 1-antitrypsin. Prenatal prognosis of 1-antitrypsin deficiency by direct evaluation of the mutation site in the gene. Comparison of the chemical bodily and survival properties of normal and Z-variant 1-antitrypsins. Repair of the secretion defect in the Z form of 1-antitrypsin by addition of a second mutation. Degradation of a mutant secretory protein 1-antitrypsin Z within the endoplasmic reticulum requires proteosome activity. Antielastases of the human alveolar buildings: Implications for the protease-antiprotease concept of emphysema. The protease-antiprotease steadiness within the human lung: Implications for the pathogenesis of emphysema. Successful spleno-renal shunt and splenectomy in two sufferers with 1-antitrypsin deficiency. Biochemical efficacy and safety of monthly augmentation remedy of alpha-1antitrypsin deficiency. Strategies for aerosol therapy of alpha-1-antitrypsin deficiency by the aerosol route. Psychological penalties of neonatal screening for alpha1-antitrypsin deficiency. They originated from an endosymbiotic alphaproteobacterium of the genus Rickettsia, which was internalized by the ancestor of all eukaryotes (1). Apart from power production, mitochondria carry out essential steps of heme, iron-sulfur cluster, and amino acid biosynthesis in addition to fatty acid oxidation and play an important function in calcium homeostasis and cellautonomous innate immunity (2). Along with innate immune signaling and apoptosis, the extremely dynamic morphology of mitochondria is one of the traits of the organelle that clearly differentiate it from most bacteria. Mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) coordinate outer membrane fusion by homo- and heterotypic interactions, whereas optic atrophy 1 (Opa1) mediates fusion of the inside membrane. Modulation of mitochondrial capabilities can be advantageous for bacteria in phrases of entry to nutrients and/or evasion of the humoral immune system. Here, we explore the connection between intracellular micro organism and host cell mitochondria, primarily focusing on the impact of the micro organism on mitochondrial morphology and manipulation of host cell dying. Manipulation of cell dying permits the micro organism to both preserve their intracellular area of interest by enhancing survival of the host cell or favor dissemination by inducing host cell demise. We current examples of each cytosolic and intravacuolar pathogenic micro organism, together with Listeria monocytogenes, Shigella flexneri, Rickettsia spp. While cytosolic micro organism are able to immediately work together with mitochondria and different organelles, intravacuolar pathogens are confined within a membrane-enclosed vacuole and employ specialized secretion systems to introduce effector proteins into the host cell cytoplasm that target mitochondria. Whether mitochondrial fragmentation directly impacts the host cell metabolic swap to glycolysis (12) stays speculative. After crossing the colonic epithelium, the facultative intracellular pathogen infects both myeloid immune cells and intestinal epithelial cells. The bacterium then quickly escapes from the phagosome and proliferates in the cytosol, where it employs the host cell actin equipment for intracellular motility in addition to for cell-to-cell spread (14). Interestingly, mitochondria have been observed at bacterial invasion websites and appear to be entrapped in an actin meshwork induced by the bacterium (15). Another examine reported local mitochondrial fragmentation within the context of counteracting host cell defense by way of septin cages. Septin cages have been described to cut back an infection by actin-polymerizing bacteria, focusing on them to autophagosomes, thus limiting both their motility and dissemination (17). In epithelial cells, necrosis is counterbalanced by the bacterium, inducing Nod1 signaling. Rickettsia species are Gram-negative obligate intracellular bacteria and are additional categorised into two main antigenically outlined teams, the typhus group and the spotted fever group. Rickettsia prowazekii represents the prototype of the typhus group and is transmitted by lice to humans, causing epidemic typhus. Rickettsia rickettsii and Rickettsia conorii belong to the noticed fever group and are the causative agents of Rocky Mountain spotted fever and Mediterranean noticed fever, respectively. After coming into the host cell via induced phagocytosis, the bacteria quickly escape the phagosome and replicate within the cytoplasm (21, 22). The authors advised that a primitive protein import mechanism hijacking mitochondrial proteins underlies the obligate endosymbiotic way of life of rickettsiae (23, 24). By regulating ranges and localization of pro- and antiapoptotic Bcl2-family proteins, R. The secreted bacterial effectors SdhA (34) and SidF have been later shown to forestall apoptosis. By stopping the fusion of the phagosome with lysosomes and inhibiting phagosomal acidification, M. The initiation of apoptosis is subsequently thought of a defense technique of host cells to restrict M. Chen and colleagues correlated the primarily induced cell demise in macrophages with mitochondrial membrane perturbations. As a consequence, the virulent strain rapidly triggers necrosis, whereas the avirulent strain induces apoptosis solely 48 h after an infection (42).
Periwinkle (Madagascar Periwinkle). Slip Inn.
- Diabetes, cancer, fluid retention, cough, lung congestion, sore throat, eye irritation, skin infections, and other conditions.
- Dosing considerations for Madagascar Periwinkle.
- Are there any interactions with medications?
- How does Madagascar Periwinkle work?
- Are there safety concerns?
- What is Madagascar Periwinkle?
Source: http://www.rxlist.com/script/main/art.asp?articlekey=96629

Order slip inn 1pack overnight delivery
In nine sufferers herbals for hot flashes 1pack slip inn purchase amex, the delay was between one and 6 years after the onset of signs [3] herbals ltd 1pack slip inn order with mastercard, and on this time, all developed cardiomyopathy, and all but one had muscle weak spot. In some sufferers, mild muscle weak point may not have been noted because of the eye devoted to the main cardiac manifestations. The advent of newborn screening for this disease made it clear that it may additionally be asymptomatic properly into grownup life [14�16]. Confronted with an irregular C0-screen, all of us now examine the mother as properly as the baby. In 15 reported households by which the defective maternal transporter was confirmed by studies of uptake of carnitine by fibroblasts, mutational evaluation, or each, the infant was normal in all but one; in that household, mom and infant were affected. Three had low stamina, simple fatigability with exercise and fasting intolerance [16]. Asymptomatic status has been observed in a father and son with faulty fibroblast uptake of carnitine and the identical mutations as another who offered in infancy with a Reye-like syndrome of hepatic disease and encephalopathy [17]. Lack of correlation between levels of carnitine and phenotypic severity is consistent with the unpredictable function of intercurrent infection [18]. Affected siblings of each sexes have been noticed, and consanguinity has been current in a minimal of 5 families [3, 21]. Prior to new child screening, the analysis was normally suspected on the idea of a low focus of free carnitine in plasma. In 20 sufferers [3], total plasma carnitine ranged from 0 to 9 mmol/L, with 18 having values less than 4. The excretion of carnitine within the urine is inappropriately excessive, consistent with defective renal tubular reabsorption [10]. At a time when the plasma carnitine approximated zero, the renal excretion was 126 mmol/g creatinine (normal, 167�425) [1], and following a dose of a hundred mg/kg of oral carnitine, the plasma carnitine rose only to 21 mmol/L, but urinary excretion elevated to 2911 mmol/g creatinine. After four months of carnitine treatment, the plasma concentrations reached the low normal vary, whereas urinary excretion was four to 5 instances normal. The fractional excretion price for free-carnitine was nearly 100 percent of the filtered load. In control cells, the uptake of 14C-labeled carnitine was by way of a high-affinity, carrier-mediated transport process with an apparent Km of 3. Fibroblasts from patients have proven 274 Carnitine transporter deficiency little uptake of carnitine; at a concentration of carnitine of 5 mmol/L, control uptake was 0. High-affinity transport is best proven at lower concentrations; up to 1 mmol/L uptake was negligible. Uptake in patients at excessive concentrations, such as 10 or 20 mmol/L replicate a second low-affinity transporter [22] or passive diffusion [24]. The uptake of carnitine by fibroblasts at 5 mmol/L confirmed no overlap among sufferers and controls. The velocity of carnitine uptake can be measured in lymphoblasts, in addition to fibroblasts [25]. Heterozygosity can be demonstrated in some sufferers by charges beneath forty % of control. Low uptake of carnitine has additionally been demonstrated in cultured myocytes derived from patients [26]. Prenatal diagnosis has been accomplished by demonstration of defective uptake of carnitine from amniocytes of an affected fetus [27]. In response to the administration of carnitine, ranges within the liver return to regular, whereas these in muscle respond poorly, indicating that the transport defect includes muscle and kidney, however not the liver. Consistent with this, the low Km and desire for L-isomer that characterize the uptake of carnitine by fibroblasts is shared by coronary heart and muscle [23, 24], however not by the liver [28]. The protein accommodates 557 amino acids and has the properties of a high-affinity transporter. There have been a few situations of the same mutation in unrelated patients [5, 32�36]. However, decisions as to severity of phenotype often rest on whether or not hypoglycemia as quickly as occurred early in life. Differences in presentation could merely reflect the chance occurrence of an intercurrent illness that led to fasting. Among ethnic differences, an 11-bp deletion was present in unrelated sufferers from Switzerland and neighboring northern Italy [3] and R169W was present in two unrelated families in Italy [36]. In Japan, the place the disease seems relatively frequent, most families have had a couple of mutations [39]. Echocardiographic study indicated asymptomatic cardiac hypertrophy in these heterozygotes. Two Iranian Jewish siblings with the identical mutation (R399Q) had very totally different clinical displays [41]. Her older sibling had proximal limb girdle weakness, which was markedly improved following treatment with carnitine. An animal mannequin of the carnitine transporter defect, the juvenile visceral steatosis (jvs) mouse [44], has autosomal recessive fatty infiltration of the liver, hypoglycemia, and hyperammonemia two weeks after start, and very low levels of carnitine in blood and muscle, along with defective renal reabsorption of free carnitine. Treatment with carnitine corrects the abnormal expression and urea cycle enzyme activity [46]. Patients with deficiency of carnitine palmitoyl-transferase I (Chapter 37) also develop hypoketotic hypoglycemia without dicarboxylic aciduria [48]. Comparisons of alterations of plasma carnitine in various issues are proven in Table 35. Levels of 3-hydroxybutyrate rose to 2 mmol/L, higher than the free-fatty acids (1. A 12-year-old patient who died abruptly following surgery [13] had been exposed to an basically vegetarian food regimen for a while. Hypoglycemia after brief durations of fasting usually symbolize issues of carbohydrate metabolism. It often takes 15�24 hours of fasting to induce hypoglycemia in a affected person with a dysfunction of fatty acid oxidation. An particular person who by no means fasted past 12 hours would usually be protected towards this manifestation. The metabolism of fats begins with lipolysis; those patients with defective fatty acid oxidation have high ratios of free fatty acids to 3-hydroxybutyrate in blood after fasting. Once transported into cells, carnitine is esterified with acyl CoA esters, including these of fatty acids resulting from lipolysis. Impaired pores and skin fibroblast carnitine uptake in major systemic carnitine deficiency manifested by childhood carnitine-responsive cardiomyopathy. Carnitine deficiency presenting as familial cardiomyopathy: a treatable defect in carnitine transport. Carnitine transporter defect: diagnosis in asymptomatic grownup women following analysis of acylcarnitines of their newborn infants.
Buy cheap slip inn 1pack
Molecular and structural analysis of metachromatic leukodystrophy patients in Indian population herbals aps pvt ltd 1pack slip inn buy overnight delivery. Attenuated actions and structural alterations of arylsulfatase A in tissues from subjects with pseudo arylsulfatase A deficiency yak herbals pvt ltd slip inn 1pack buy discount online. Juvenile and grownup metachromatic leukodystrophy: partial restoration of arylsulfatase A (cerebroside sulfatase) exercise in inhibitors of thiol proteinases. The accumulation of cerebroside sulfates by fibroblasts in culture from sufferers with late infantile metachromatic leukodystrophy. Saposin proteins: structure, operate and role in human lysosomal storage disorders. Characterization of a mutation in a family with saposin B deficiency: a glycosylation website defect. Histochemical and biochemical research of urinary lipids in metachromatic leukodystrophy and Fabry illness. Treatment of late childish metachromatic leukodystrophy by bone marrow transplantation. White matter dysfunction and its neuropsychological correlates: a longitudinal study of a case of metachromatic leukodystrophy treated with bone marrow transplant. Presymptomatic lateinfantile metachromatic leukodystrophy treated with bone marrow transplantation. The future for therapy by bone marrow transplantation for adrenoleukodystrophy, metachromatic leukodystrophy, globoid cell leukodystrophy and Hurler syndrome. Successful treatment of metachromatic leukodystrophy utilizing bone marrow transplantation of HoxB4 overexpressing cells. Unrelated umbilical wire blood transplant for juvenile metachromatic leukodystrophy: a 5-year follow-up in three affected siblings. Enzyme alternative improves nervous system pathology and function in a mouse model for metachromatic leukodystrophy. This post-translational change affects the whole sulfatase household, a minimum of seven members of that are lysosomal enzymes which are specifically involved within the degradation of sulfated glycosaminoglycans, sulfolipids, or different sulfated molecules. The gene has been recognized, and missense mutations have been discovered which lead to variable lack of perform [9]. The deficiency of steroid sulfatase is answerable for the skin lesions of X-linked ichthyosis. Among the enzymes of mucopolysaccharide metabolism: (1) deficiency of iduronate sulfatase offers manifestations of Hunter syndrome; (2) deficiency of heparan sulfatase yields the impaired psychological improvement and cerebral features of Sanfilippo A disease; (3) deficiency of N-acetylglucosamine-6-sulfatase, those of Sanfilippo B illness; (4) deficiency of N-acetylgalactosamine-6-sulfatase offers rise to options of Morquio disease; and (5) deficiency of N-acetylgalactosamine-4-sulfatase, also referred to as arylsulfatase B, causes features of Maroteaux-Lamy disease, together with corneal clouding. Obviously, completely different degrees of deficiency or quantities of residual enzyme activity can be expected to result in quite completely different phenotypes. A variety of totally different clinical phenotypes have been delineated [4], together with the basic late infantile kind, a neonatal kind, a juvenile form, and a Saudi variant. Death might occur at 10�18 years, however there was survival into the third decade. Mucopolysaccharidosis-like features possibly evident very early in life [12, 18], and may well be the first evidence of illness. The prognosis ought to be thought-about in young patients with signs of mucopolysaccharidosis. Virtually all sufferers have roentgenographic evidence of dysostosis multiplex (Chapters 76 and 78). Ophthalmologic findings have included optic atrophy, retinal degeneration, and nystagmus [5, 10, 19]. A milder juvenile kind with onset at about five years of age has been reported [6]. Early development could additionally be normal, and sufferers may walk and speak at regular instances [4], however some may be developmentally delayed early on. Increased deep tendon reflexes and ankle clonus may be followed by spastic quadriparesis. Facial features had been coarse, the nasal bridge depressed, and the nasal tip tilted, highlighting the ample nasal subcutaneous tissue. Consistent with the severity of the phenotype, enzyme activities of all the sulfatases examined have been very severely depressed [12, 21]. Abnormal teeth have been reported [22] in a lady who died at thirteen, and have been proposed as an early (potentially at birth) diagnostic software. The tooth have sharp cusps, thin hypomineralized or missing enamel and exposed discolored dentin. A Saudi variant has been distinguished [4] during which there was early infantile onset of extreme dysostosis multiplex, showing as Maroteaux-Lamy syndrome or Morquio illness. Deafness was absent, but one patient had abnormal auditory evoked potentials on one aspect. Ichthyosis was absent, and in six of seven sufferers studied the activity of steroid sulfatase was regular [4]. Patients had gentle to reasonably impaired mental improvement, and one patient had a normal cognitive quotient despite motor impairment. Most of these sufferers had retinal adjustments, however two had lenticular opacities, which have been seen, but hardly ever within the traditional presentation [5]. Two sufferers had proof of cervical twine compression with the development of sudden quadriparesis, followed in one by death. In the Saudi patients, premature synostosis of a quantity of cranial sutures led to deformities, such as trigonocephaly, brachycephaly, or dolichocephaly [4]. Abnormalities of the odontoid have been observed [4]; C1 has been lower than normal. A four-year-old girl with microcephaly had neurologic regression and spondylolisthesis, however no ichthyosis or coarse facies [24]. Flexion of the hips and knees, in addition to the elbows contributed to the Morquio-like appearance. Laboratory findings in all these patients embody mucopolysacchariduria (dermatan sulfate and heparan sulfate). Diagnosis is made by confirming the deficiency within the activity of a variety of sulfatase enzymes [12]. Newborn screening for lysosomal storage illnesses has been developed through quantification of immunoreactive lysosomal proteins [26]. A localized fairly marked constriction of the anteroposterior diameter led to compression of the wire to about one-half normal size. Moderately intensive excessive T2 adjustments within the subcortical white matter have been present bilaterally. Both sexes have been equally represented, often with multiple affected patient in a sibship. Defective activity of sulfatases could be proven in cultured fibroblasts [18, 19] and in tissues, corresponding to kidney, brain, and liver [2, 12, 31]. There has been some correlation of the degrees of residual enzyme exercise and scientific phenotype [3, 29, 32].
1pack slip inn buy mastercard
This byproduct of galactose accumulation occurs by its discount at carbon-1 and is current in urine and tissues herbals himalaya purchase slip inn 1pack amex. In addition herbals plant actions discount 1pack slip inn otc, cataracts that result from galactose treatment of rats are prevented by sorbinil, which inhibits aldose reductase, the enzyme that catalyzes the conversion of galactose to galactitol [74]. Galactitol has been demonstrated in vivo by proton magnetic resonance spectroscopy within the mind of an encephalopathic toddler with galactosemia [75]. Information on mutations has indicated that the Q188R/ Q188R genotype is a major predictor of developmental verbal dyspraxia in sufferers with good metabolic control as indicated by erythrocyte Gal-1-P levels less than 3. Q188R mutation and one who had severely impaired mental improvement had the often milder p. Studies of glycosylation of N-linked glycoproteins revealed persistent aberrant glycosylation. The mainstay of the diet for an toddler is the substitution of casein hydrolysate or a soybean preparation for milk formulation. Education of the parents and of the kid as she or he grows older on the galactose content material of meals is essential. The willpower of the Gal-1-P content material of erythrocytes is employed in monitoring adherence to the food regimen [81], and acceptable levels have been set at four mg/dL (150 mmol/L). Experience with early therapy helps the concept that effective therapy instituted in the first weeks of life can forestall many of the traditional manifestations of the illness. At the opposite end of the scale, impaired psychological development, once established, is irreversible, and if the diagnosis is delayed, some injury to the brain is inevitable. There may be abnormalities of visible perception, behavior issues, or convulsions. Cataracts are reversible if therapy is began within the first three months of life. A recent report of long-term follow up of 28 patients handled for traditional galactosemia confirmed imply beneath common operate across a broad spectrum of cognitive measures [82]. However, there was a variety, and some people had common or above common success. However, plasma concentrations of galactose and galactitol were about twice that of control. Supplemental calcium and vitamin D should be provided, and serum 25-hydroxyvitamin monitored at least yearly. The enzymatic transformation of uridine diphosphate glucose into a galactose spinoff. The construction of the galactose-1-phosphate current within the liver throughout galactose assimilation. Hypergonadotropic hypogonadism found already in pre-pubertal women however only in adult males. Granulocyte function in patients with galactose-1-phosphate uridyltransferase deficiency (galactosemia). Computerised tomographic demonstration of cerebral edema in a toddler with galactosemia. Biogenesis of galactose, a attainable mechanism of self-intoxication in galactosemia. Purification of normal and inactive galactosemic galactose-1-phosphate uridyl-transferase from human purple cells. The enzymatic expression of heterozygosity in households of youngsters with galactosemia. Screening for galactosemia and glucose6-phosphate-dehydrogenase deficiency in new child infants. A new genetic abnormality resulting in galactose-1-phosphate uridyltransferase deficiency. A new variant of galactose1-phosphate uridyltransferase in man: the Los Angeles variant. An improved electrophoretic process for galactose-1-phosphate uridyl transferase: demonstration of multiple exercise bands with the Duarte variant. Galactose-1-phosphate uridyltransferase deficiency because of Duarte/galactosemia mixed variation: Clinical and biochemical studies. Hereditary galactokinase deficiency a newly recognized reason for juvenile cataracts. Galactose cataract prevention with sorbinil and aldose reductase inhibitor: a light microscopic examine. In vivo evidence of mind galactitol accumulation in an toddler with galactosemia and encephalopathy. Correlation of ovarian with galactose-1-phosphate uridyl transferase ranges in galactosemia. Alpha-D-galactose-1-phosphate willpower as galactose after hydrolysis of phosphate. A pilot examine of biochemical and neurodevelopmental evaluation in children detected by new child screening. Monitoring of biochemical standing in children with Duarte galactosemia: utility of galactose, galactitol, galactonate, and galactose 1-phosphate. Newborn screening for galactosemia in the United States: Looking back, looking around and looking forward. Assignment of the human gene for galactose-1-phosphate uridyltransferase to chromosome 9: research with Chinese hamster-human somatic cell hybrids. Regional localization of human gene loci on chromosome 9: studies of somatic cell hybrids containing human translocations. Origins, distribution and expression of the Duarte-2 (D2) allele of galactose-1phosphate uridylyltransferase. Galactosemia: a technique to establish new biochemical phenotypes and molecular genotypes. Identification of galactose-1phosphate uridyltransferase geen common mutations in dried blood spots. They are a heterogeneous group with different etiologies and totally different scientific manifestations. The traditional form of glycogen storage disease was first described by von Gierke in 1929 [1]. The glycogen from this authentic affected person was isolated by Sch�nheimer [2] and was discovered not to differ from normal glycogen in optical rotation or in its composition of glucose residues. This seems to have been the primary demonstration of the concept proposed by Garrod [3] that inborn errors of metabolism outcome from genetically determined deficiencies of single enzymes. Glycogen is a branched, polydisperse molecule that has been acknowledged since the time of Claude Bernard because the storage kind for carbohydrates in animal tissues. The straight chains of glucose residues are linked collectively by -1,4 bonds; branching occurs by way of 1,6 linkages. The predominant structure is that of straight chains of glucose molecules in 1,4 linkage.

Discount 1pack slip inn otc
The outermost layer herbalshopcom buy generic slip inn 1pack line, the stratum corneum herbs list order slip inn 1pack overnight delivery, consists of tightly packed cornified squamous epithelial cells which are produced by the constantly dividing keratinocytes and constantly replenishing the barrier against the useless cells and microbes. This layer additionally acts as a barrier in preventing dehydration and limiting entry of microbes through the pores and skin. The clean translucent layer of the dermis stratum lucidum is positioned simply above the stratum granulosum and below the stratum corneum. These layers are composed of cells densely packed with the lipid eleiden, which is derived from keratohyalin; the cells have a lucid look and act a barrier to water. The stratum granulosum is filled with cells containing the fibrous proteins keratin and keratohyal and has a grainy appearance. The stratum basale or stratum germinativum is the deepest epidermal layer, and beneath it lie the layers of the dermis. Dermal papillae current on this layer improve the energy between the dermis and dermis [4]. In the respiratory tract, mucus traps inhaled micro organism, fungi, and environmental pollutants. Microbes attached to cells within the mucous membranes can be removed by the sloughing of the epithelial cells. Respiratory tract: the beating of the cilia in the epithelia lining the respiratory tract helps to propel outward the inhaled particles, and pathogens entrapped in mucus. The respiratory epithelia, if disrupted by smoke, fumes, or pollution, can evidence increased respiratory disorders [5]. In the genetic disorder, cystic fibrosis, which is due to a mutant gene-encoded defective ion channel, leads to thick and viscous secretions that obstruct the respiratory tract, leading to infections by micro organism like Pseudomonas aeruginosa [6]. Urinary tract: Urination removes microbes from the urinary tract, bladder and kidneys. The time-to-time launch of the urine prevents the upward movement of the pathogens within the urinary tract. The use of a urinary catheter disrupts this action and will increase the probabilities of an infection. The feminine genital tract is moreover protected by acidic vaginal secretions and microcidal molecules in secretions [7]. In the pores and skin, oil and sebum, which are lipid in nature, are secreted from sebaceous and sweat (or sudoriferous) glands and have an acidic pH that stops the growth of microbes. Excessive sebum can lead to oily skin with acne and clog skin pores, entrapping microbes. Secreted molecules: the innate immune cells launch microcidal molecules in response to microbial publicity that inhibit or lyse them. The lysozyme in sweat can breakdown the peptidoglycan wall of bacteria and cause bacterial cell lysis. Sweat, on evaporation, creates a salty setting that forestalls bacterial development [8,9]. In people, defensins consist of six invariant cysteines with three disulphide bonds and produce a secondary structure consisting of three strands of anti-parallel sheets. They act on the outer cell membrane surrounding the pathogen, Innate Immunity 113 2. In the respiratory tract, the defensins, secreted by the respiratory epithelium, protect the lung mucosal surfaces. They increase the susceptibility of microbes to ingestion and digestion by phagocytic cells. It is reported to be overexpressed in response to pathogens like fungi and micro organism. Along with lysozyme in the saliva, different digestive enzymes released within the mouth and by salivary glands, and gastric glands and bile and pancreatic enzymes within the small intestine can kill microbes. The lacrimal glands, situated in the outer portion of the attention, launch tears that include lysozymes that protect the eye from infectious agents. Histatins 1 and three are encoded by a gene situated on chromosome 4q13, whereas others are the cleaved merchandise of those two peptides. Dermcidin: Dermcidin, an anionic defence peptide, is constitutively expressed in human sweat. These bacteria are non-pathogenic to the host beneath normal situations, and any disruptions of this microbiome can result in illness. The use of antibiotics can cause lysis of the intestine microbes and promote the establishment of pathogenic organisms like Clostridium difficile, causing a pathological situation called pseudomembranous colitis with symptoms of watery diarrhoea, stomach pains, and fever. The plasma membrane folds inward to kind small endocytic vesicles often recognized as endosomes. Pinocytosis involves non-specific membrane folding, whereas receptor-mediated endocytosis involves selective internalisation of specific macromolecules following their binding to particular membrane receptors. After internalisation, the endosomes fuse with main lysosomes, which comprise degradative enzymes similar to proteases, nucleases, and lipases that hydrolyse the internalised macromolecules and break them down into small merchandise that are launched by exocytosis. These later fuse with lysosomes, forming phagolysosomes that carry out the digestion of the engulfed matter. Macrophages ingest exogenous antigens, together with microorganisms and insoluble proteins and endogenous antigens including lifeless or injured host cells. Phagocytosis initiates when antigen adheres to the macrophage membrane, which then sends out pseudopodia-like structures around the target object and varieties a membrane-bound structure referred to as a phagosome. The phagosome is then endocytosed and is moved into the cell interior, where it fuses with a lysosome after which is identified as a phagolysosome. Lysosomes contain a wide range of hydrolytic enzymes, digest the ingested material, and then eliminate the remnants by exocytosis. The macrophage membrane has receptors for antibodies and may bind to antibody-coated antigen and improve phagocytosis. This course of is named opsonisation and is where the antibody capabilities because the opsonin, binding the antigen to the phagocyte and enhancing phagocytosis. These macromolecules include adaptor molecules, kinases, and transcription components, which might result in the expression of pro-inflammatory molecules, cytokines, chemokines, cell adhesion molecules, and immunoreceptors that then allow the early host responses to an infection and set off adaptive immunity [2]. Downstream signalling and gene activation lead to up-regulated expression of cytokines, chemokines, and immune effector molecules. Signalling cascades initiated by cytokines and chemokines have been identified to play main roles in cellular development and growth, haematopoiesis, lymphocyte recruitment, T-cell subset differentiation, and inflammation. They show a similar four helical region by which the primary and second helices and second and fourth helices are parallel to one another and interconnected by loops that predominantly act regionally. The type I cytokine receptor household incorporates conserved motifs of their extracellular amino acid domain and lacks signalling motifs. They also induce macrophages and endothelial cells to launch chemokines and induce leukocyte chemotaxis and infiltration. Secreted in giant portions, it could possibly cause myocardial contractions, inhibition of vascular smooth muscular tissues, intravascular thrombosis, metabolic disturbances, and promote septic shock from bacterial endotoxins. They promote clonal growth and induces fever and the synthesis of acute phase proteins. They improve the floor expression of ligands on endothelial cell surfaces, enabling leukocyte adhesion.
1pack slip inn generic mastercard
Psychomotor improvement was normal for two months; weak point of neck muscular tissues was first discovered at three weeks herbals in the philippines slip inn 1pack purchase overnight delivery. Nonclassic or late-onset forms of K rabbe galactosylceramide lipidosis have been recognized increasingly for the rationale that advent of enzymatic diagnosis [30� 33] herbals export slip inn 1pack generic. Most have introduced by ten years of age, however in others neurologic signs developed between ten and 20 years, and one was reported at 39 years of age [31, 34, 35]. In the late infantile group of patients in whom the onset was between six months and three years [36], the manifestations and development have been little completely different from the classic disease, and dying often ensued within two years of onset. In a second group, in whom the onset was three to eight years [36], the progression was slower, and none had died within the period of observe up, which was so long as seven years. Some were developmentally delayed earlier than the onset of degradation [32, 37]; some had seizures [38�40]; and two had hemiparesis, progressive in a single to tetraplegia [41]. Adult patients have been described in whom onset was between 10 and 35 years of age. Adultonset sufferers are being more and more reported [33, 43] with progressive spastic paraparesis or peripheral neuropathy. In basic Krabbe illness and its variants, neuroimaging normally signifies diffuse cerebral atrophy [44�46]. Plaque-like excessive depth T2 signal has been noticed in periventricular and cerebellar white matter in three sufferers [49]. In 82 % of 27 patients, at some point to eight years old, there was uniform slowing of sensory and motor nerve condition [50]. The patient could have hyperactive deep tendon reflexes, while electrophysiologic studies point out a distinguished peripheral neuropathy [24]. The neuroanatomic pathology of Krabbe disease is characterised by an extreme hardness or sclerosis of the white matter. These massive irregular cells range from 20 to 50 microns in diameter and comprise as many as 20 nuclei. The ultrastructure of the globoid cells reveals irregular tubular crystalloid inclusions [14, 19, 21, 54]. The identical inclusions in globoid cells have been produced in rats by intracerebral injection of galactocerebroside [55]. These observations advised that the cells accumulate galactosylceramide, and this has been documented by chemical evaluation [2]. The disease seems to be widespread in Scandinavia; the incidence in Sweden was calculated to be 1. It is now possible by enzymatic assay of chorionic villus material, in addition to amniocytes. It is really helpful that enzyme assay be carried out on dad and mom earlier than prenatal analysis is undertaken to avoid a false positive in the case of a very low worth in a heterozygote. Once the mutation is thought, molecular analysis could also be carried out for heterozygosity and prenatal diagnosis. Cerebrosides are monohexosyl ceramides by which the sugar is glycosidically linked to the C-1 of ceramide. Galactosylceramide is the attribute cerebroside of myelin and of the central nervous system. The compound is often degraded to ceramide and galactose by the lysosomal enzyme galactosylceramide -galactosidase [6]. In sufferers, the extent of exercise has been documented to be 5�10 p.c normal in mind, liver, spleen, and kidney [6, 7]. The assay is conveniently and reliably carried out on leukocytes or cultured fibroblasts [59]. Enzymatic diagnosis with the natural substrate is demanding and must be carried out in an skilled laboratory [57, 60]. A mutant allele has been reported [61] in which the galactosidase exercise overlaps that of patients with Krabbe illness. The proband of the first family was a wholesome public health nurse who had volunteered as a control in a research of Krabbe illness. The presence of this new allelic gene might result in a misdiagnosis of Krabbe disease, especially in utero. The situation might be like that of the Duarte variant for galactose-1phosphateuridyltransferase, in which compound variants have been noticed who were heterozygous for both the gene for galactosemia and that for the Duarte variant. These findings reinforce the advice to establish the enzymatic profile in parents earlier than enterprise a prenatal diagnosis. Methodology has been developed for dried blood spots during which the product is assayed by tandem mass spectrometry, which allows newborn screening [62�64]. The twitcher mouse has an autosomally recessively determined deficiency of galactosylceramide -galactosidase and is an interesting mannequin for Krabbe disease [65]. Other fashions have been found in West Highland and Cairn terriers, sheep, and monkeys. T1637C, which reduces activity slightly, is found on one allele and a disease-causing mutation, such as c. I66M and I289V, which have been found only in Japanese, accounted for 37 mutant alleles, and with p. It is an uncommon lipid storage disease, in that the stored substrate accumulates only in globoid cells. The disease in the mouse differs in that inclusions are seen, and the cerebroside accumulates in each kidney and lymphocytes. In both mouse and man, ranges of psychosine had been elevated in brain and peripheral nerves [70, 71]. The terminal galactose is cleaved from this 710 Krabbe disease/galactosylceramide lipidosis/globoid cell leukodystrophy 11. Cherry red spot in affiliation with galactosylceramide-beta-galactosidase deficiency. A new mutation in an infant with Krabbe illness accompanied by enlargement of the optic nerves. Late onset Krabbe disease (globoid cell leukodystrophy): clinical and biochemical options of 15 instances. Oligodendroglia seem to be selectively destroyed by psychosine fashioned within them. Among eleven infantile-onset sort patients given stem cell transplantation before the onset of symptoms, they were reported [76] to have normal ranges of galactocerebroside in blood, progressive myelination and regular cognitive operate in most, however some had delayed improvement. The cloning of the gene and the supply of animal models present avenues for the research of gene remedy [77]. Late-onset globoid leukodystrophy: uncommon ultrastructural pathology and subtotal beta-galactocerebrosidase deficiency. Further observations on the nice construction of globoid leukodystrophy: peripheral neuropathy and optic nerve involvement. Pilot examine of new child screening for six lysosomal storage disease utilizing Tandem Mass Spectrometry. Clinical outcomes of children with abnormal new child screening results for Krabbe illness in New York State. Cellular response of the brain to exogenous galactosylsphingosine monogalactosyl diglyceride and lactosylceramide.